Session Chairs: Laurie Krug & Bettie Steinberg
Circular RNAs
Y Chang, N Lee3, PS Moore1,3, MA Nalesnik2, S Ranganathan2, KH Shair3, SH Swerdlow2, Tuna Toptan1,2, J Wendzicki1
1Hillman Cancer Center, 2University of Pittsburgh Medical Center, 3Department of Microbiology and Molecular Genetics
CircRNAs are formed by backsplicing of linear RNA and can be exonic and/or intronic. Due to their covalently joined circular nature, circRNAs are resistant to exonucleases and therefore highly stable. To date no circRNAs have been described from DNA tumor viruses. We performed RNA sequencing on exonuclease (RNaseR) treated EBV positive and negative post-transplant lymphoproliferative disease (PTLD) patient samples and identified EBV backspliced junction reads within the RPMS1 locus only in EBV positive PTLD. These junction reads were determined to be from circular RNAs by RT-PCR using divergent primers. They comprise 4 differentially spliced isoforms that vary in size based on specific combinations of intronic and exonic elements. In extended sampling, these viral circRNAs were found in all 8 EBV+ but none of 9 EBV- PTLD tested. We also verified the presence of circRPMS1 in all EBV+ cell lines tested, except in the B95-8 line, which harbors a deletion in this locus. CircRPMS1 forms are present during both latent and lytic replication, although we detected some variation in relative quantity from cell line to cell line. Furthermore, circRNAseq analysis of KSHV-infected PEL cell lines revealed several species of KSHV circRNAs, which can also be detected in KS lesions. Gammaherpesviruses generate circRNAs as a novel class of viral transcripts.
See notes ^o^ (Pat Moore and Yuan Chang Lab)

Takanobu Tagawa1, Joseph Ziegelbauer1
1HIV and AIDS Malignancy Branch, National Institute of Health
Non-coding RNAs have wide implications in host-virus interactions; Herpesvirus-encoded miRNAs reduce apoptosis, promote cell cycle and growth, and reduce immunogenicity; PAN RNA in KSHV and EBERs in EBV are involved in successful viral infection. Circular RNAs (circRNA) are novel single-stranded non-coding RNAs with long half-lives, sequence conservation, localization in cytoplasm, and abundant expression. CircRNAs are products of back-splicing and potential to antagonize RNA binding proteins, mRNAs, and miRNAs in sequence-dependent manner. Human circRNAs have been shown to inhibit human miRNAs.
We hypothesized that circRNAs influence viral infection by inhibiting host and/or viral factors. Viral miRNAs are particularly attractive targets of circRNAs since viral miRNAs are produced by same mechanism of human miRNAs and less likely to be distinguished through synthetic pathway. We found that many human circRNAs are enriched with predicted viral miRNA target sites. Screening with RNA-Seq analysis of KSHV-infected Human Umbilical Vein Endothelial Cells (HUVEC) identified hundreds of human circRNAs that are differentially regulated upon infection. We confirmed the expression changes with RT-qPCR and RNase R treatment. We are pursuing gain and loss of specific circRNAs to investigate their effects on infection.
Furthermore, we examined whether herpesviruses express their own circRNAs as in the case of viral miRNAs. We have identified multiple KSHV circRNAs expressed in KSHV- infected endothelial cells and primary effusion lymphoma cells. Upon the induction of lytic cycle, some viral circRNAs are up-regulated. To our knowledge, circRNAs have not yet been reported for sub-genomic viral RNAs.
We are continuing to investigate the functional roles of human and viral circRNAs in inhibiting or benefitting infection. Both miRNAs and circRNAs are by nature non- immunogenic and long-lived compared to conventional linear RNA molecules. This research has revealed new host-viral interactions with circRNAs that may play key roles for successful infection by herpesvirus.
Circular RNA
- Product of back splicing
- Resistant to ending lease
- Identifying back-spploiced junctions
- Divergent primer PCR
- Resistant to RNase R
Does KSHV cause differential expression of human circRNAs? (Hsa_circ_0001400 is the highest one unregulated by KSHV infection)
- KSHV infection up-regulation specific human circRNAs in endothelial cells
- SLK 3 days post infection
- Hsa_circ_0001400 suppresses latent and lytic gene expressions
- RTA
- But viral genome numbers are not affected
Does KSHV encode CircRNA
- Arise from lytic gene locus in unfected HUVECs
- PAN and ORF56!!
Viral circRNAs correlate with RTA expression in patients
Lineage transcripts
- LANA
- RTA
- Kcirc3, 29, 57
Viral circRNA reduces RTA, but not LANA, gene expression in SLK cellsViral circRNAs can regulate cell growth: kcirc54
Potentials of KSHV circular RNAs
- Decoy of antiviral RNAs and RBPs
- Long-lasting and cost effective
- Adaptive immune evasion
- Refjuction of lytic gene expression
- Spread via extracellular vehicles
- KSHV circRNAs May avoid recognition by RIG-1 (Chen et al, 2017)
PTPN14 to Limit Keratinocyte Differentiation
Joshua Hatterschide1, Amelia E. Bohidar1, Elizabeth A. White1
1University of Pennsylvania Perelman School of Medicine
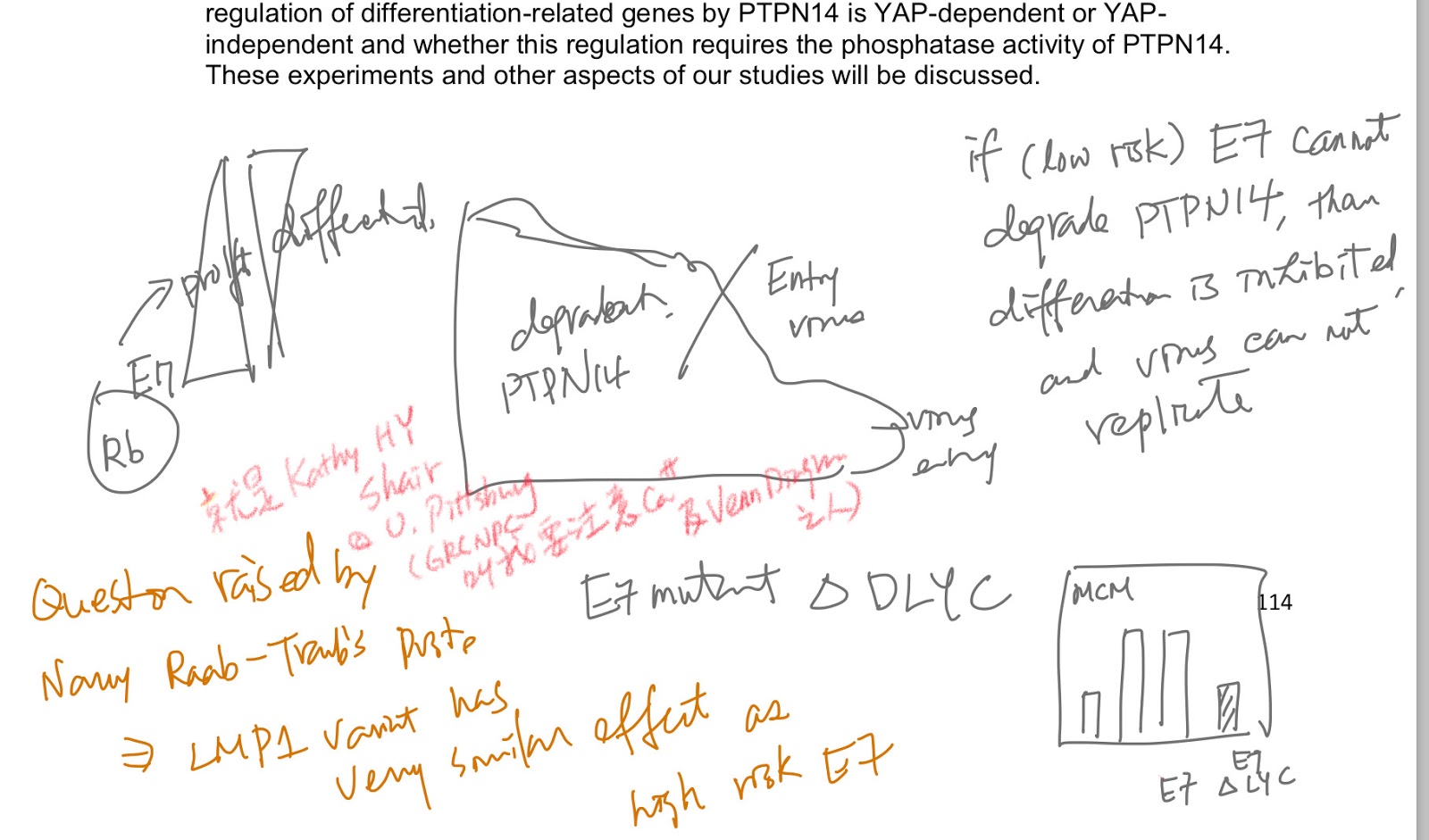
High-risk human papillomavirus (HPV) E7 oncoproteins have a transformation activity that is independent of their binding to the retinoblastoma tumor suppressor (RB1), but the molecular basis of this activity remains uncertain. We have previously determined that PTPN14, a non-receptor protein tyrosine phosphatase and putative tumor suppressor, interacts with many HPV E7 proteins. Oncogenic HPV E7 form a complex that includes the E3 ligase UBR4 and PTPN14 and use UBR4 to target PTPN14 for proteasome-mediated degradation. The ability of E7 to bind PTPN14 is correlated with its oncogenic activity in several cellular transformation assays.
We have continued to investigate the effects of PTPN14 degradation by E7 and the effects of PTPN14 knockdown or knockout in the absence of E7. We have characterized an HPV16 E7 variant that cannot bind to UBR4 but that retains the ability to inactivate RB1. Using this variant in primary keratinocyte immortalization assays has enabled us to separate the pro-proliferative effect of RB1 inactivation by HPV16 E7 from the effect of PTPN14 degradation. Gene expression analyses in cells producing HPV16 E7 or an E7 variant, or in cells depleted of PTPN14, suggest that PTPN14 promotes the expression of keratinocyte differentiation genes. We hypothesize that the degradation of PTPN14 by high-risk HPV E7 limits keratinocyte differentiation, and we are testing whether this is related to the RB1-independent transformation activity of high-risk HPV E7.
PTPN14 has been characterized as a negative regulator of YAP in the Hippo signaling pathway, but we find that a reduction in PTPN14 levels does not cause activation of pro- proliferative YAP target genes in human keratinocytes. We are testing whether the regulation of differentiation-related genes by PTPN14 is YAP-dependent or YAP- independent and whether this regulation requires the phosphatase activity of PTPN14. These experiments and other aspects of our studies will be discussed.
High-risk E7 enable proteasome-mediated degradation of PTPN14
PTPN14-E7-UBR4
UBR4 required for transformation by E7
Allows E7 to inhibit apoptosis following detachment (?)
PTPN14
- Non receptor tyrosine phosphatase
- Tumor suppressor in colorectal other cancers
- Relapse neuroblastoma
- Skin basal cell carcinoma
- PDAC
In MCF10A cells
PTPN14/YAP/TEAD
Can’t reproduce the experiments, went to YAP expert, the YAP expert said: the situation got to be complicated XDD
Oral Talk #115
Barr Virus Lytic Life Cycle in Immortalized Oral Keratinocytes
Kathleen Makielski1, Denis Lee1, Mitchell Hayes1, Bill Sugden1, Paul F. Lambert1
1University of Wisconsin-Madison
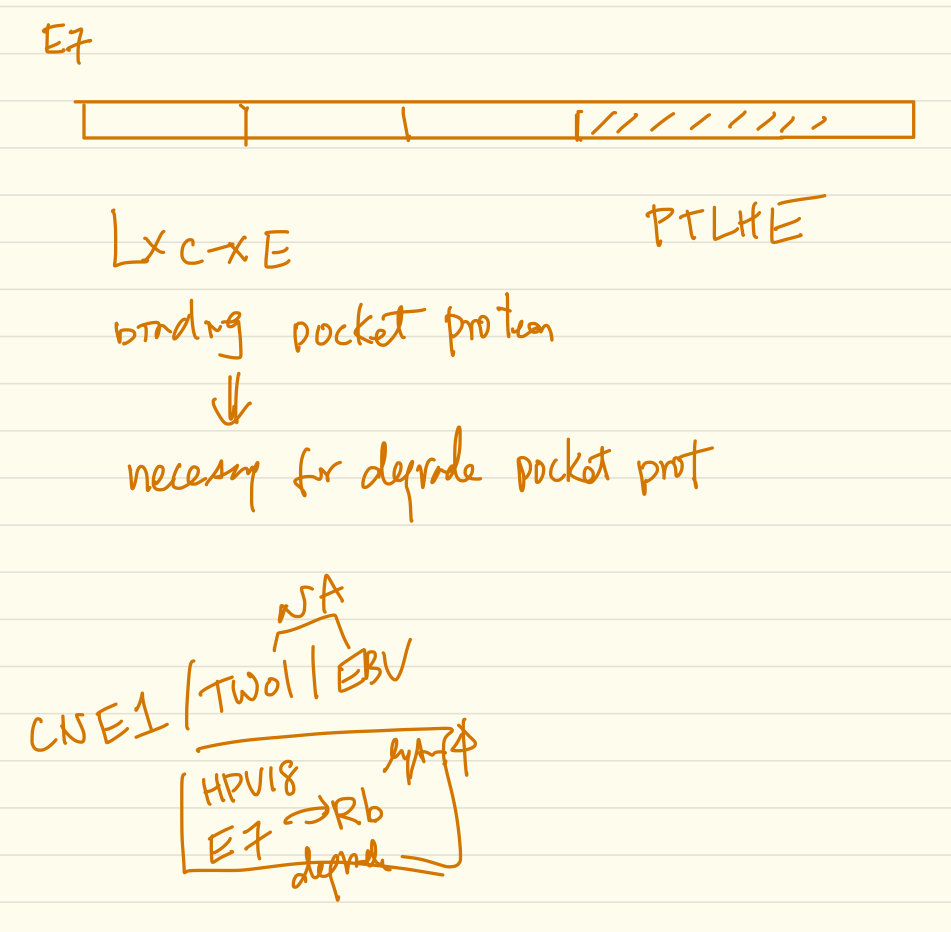
Epstein-Barr virus (EBV) and human papillomaviruses (HPVs) are human tumor viruses that cause head and neck cancers. Both viruses infect and replicate in upper aerodigestive tract epithelia. Some studies have detected co-infection in both oropharyngeal and nasopharyngeal cancers raising the possibility that these viruses could potentially affect each other’s life cycles and/or oncogenic potential. Our lab has established an in vitro model system using organotypic raft cultures to test the effects of EBV and HPV on each other in stratified squamous oral epithelial cells. We found that the presence of HPV promotes EBV’s lytic life cycle in suprabasal layers of the oral epithelium. Specifically, HPV promotes expression of an EBV immediate-early protein, Z, EBV genome amplification, and EBV infectious virus production in raft cultures. HPV’s effects on EBV’s lytic life cycle correlate with the ability of the high-risk HPV oncoprotein, E7, to bind pocket proteins. However, E7’s ability to degrade pocket proteins as well as E7’s ability to specifically bind Rb are not necessary for its promotion of the EBV lytic life cycle. Ongoing studies designed to reveal which pocket protein(s) are critical targets in mediating HPV E7 oncoprotein’s effect on the EBV life cycle will shed further light on what controls the lytic phase of the EBV life cycle in human oral epithelial cells.
Virology. 2016 Aug;495:52-62. doi: 10.1016/j.virol.2016.05.005. Epub 2016 May 11.
Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes.
Abstract

Oral Talk #116
Papillomavirus-Immortalized Keratinocytes
Joseph T Guidry1,2,3, Julia E Myers1,2, William K Songock1,2, Malgorzata Bienkowska-Haba1,2, Xiaohui Ma3, Cherie-Ann O Nathan3,4, Martin Sapp1,2,3, Jason M Bodily1,2,3, Rona S Scott1,2,3 1Department of Microbiology and Immunology, LSU Health Sciences Center, 2Center for Molecular and Tumor Virology, LSU Health Sciences Center, 3Feist-Weiller Cancer Center, LSU Health Sciences Center, 4Department of Otolaryngology, LSU Health Sciences Center
The detection of the oncogenic herpesvirus Epstein-Barr virus (EBV) in human papillomavirus-associated oropharyngeal squamous cell carcinoma (HPV+OSCC) implies a possible cofactorial role for EBV alongside HPV in OSCC pathogenesis. As EBV- associated carcinomas exhibit latent EBV infection, we hypothesized that HPV may delay epithelial differentiation required for EBV replication and promote EBV latency. To test our hypothesis, HPV16-transfected tonsillar keratinocytes grown in organotypic raft culture were infected via co-culture with EBV-positive Akata Burkitt’s lymphoma cells. Six days post-EBV infection, robust EBV replication was observed in HPV-negative tonsillar rafts, with little to no replication in HPV16-immortalized rafts. The reduction in EBV replication was independent of immortalization as human telomerase-immortalized normal oral keratinocytes supported robust EBV replication in raft culture. To identify the HPV factors responsible, human foreskin keratinocytes (HFK) expressing HPV16 oncogenes E6 and/or E7 showed that HPV16 E7 was sufficient to reduce EBV replication. EBV is dependent upon epithelial differentiation and the differentiation-dependent expression of the cellular transcription factor KLF4 for replication. While KLF4 RNA and protein levels were unaltered in HPV16-transfected or E6/E7-expressing rafts, several KLF4 cellular targets, including differentiation markers filaggrin and loricrin, were reduced. However, expression of the EBV immediate early genes, viral targets transcriptionally activated by KLF4, was not affected by HPV16. Instead, a block in EBV early gene expression was observed. These results suggest that HPV interference with EBV replication was independent of KLF4 activity on viral gene expression. Instead, HPV16 E7 interactions with retinoblastoma pocket proteins may result in a cellular reprogramming that is no longer conducive for EBV replication. HPV16 E7-mediated interruption of EBV replication may facilitate a latent EBV infection which would provide additional viral oncogene expression and contribute to the rapid progression of HPV+ OSCC.
OPC, 90% OPC occur in tonsils
PLoS Pathog. 2015 Oct 2;11(10):e1005195. doi: 10.1371/journal.ppat.1005195. eCollection 2015 Oct.
Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells.
Abstract

Oral Talk #117
Cellular Epigenome
Lena Lupey – Green1, Lisa Caruso1, Kayla Martin1, Michael Hulse1, Jozef Madzo1, Sarah Johnson1, Italo Tempera1,
1Fels Institute for Cancer Research – School of Medicine Temple University
EBV establishes a latent infection with complex and dynamic gene expression patterns. These gene expression patterns can adapt to diverse host cell types and immunological responses. The changes in viral gene expression are regulated by both viral and cellular factors, including epigenetic modulators. We previously discovered that an important epigenetic feature of EBV is the three-dimensional (3D) structure of the viral chromosome. Changes in EBV 3D chromatin structure correlates with different patterns of gene expression. The primary factor that mediates the 3D genome structure in EBV is CTCF. Our recent work reveals that EBV activates PARP1, which has been shown to be important in regulating CTCF chromatin loop formation. We performed ChIPseq in EBV+ cells to assess PARP1 binding across the EBV genome. We identified numerous PARP1 binding sites, including OriP and the active Cp promoter. We also identified numerous sites at which PARP1 and CTCF colocalize, including the Cp promoter. ChIPseq in EBV+ cells treated with and without the PARP inhibitor olaparib revealed some changes in CTCF binding across the genome, including at Cp, where CTCF was evicted from the region. By FAIRE-seq and ChIP-seq we determined that changes in CTCF binding correlate with changes in chromatin accessibility and accumulation of repressive epigenetic marks. RNA- seq analysis confirmed that these changes in the chromatin structure correlate with changes in viral gene expression. PARP1 and CTCF also regulate cellular chromatin, therefore we determined whether EBV can alter host gene expression through PARP1 in a similar way. We found that EBV activation of PARP1 changes PARP1 and CTCF binding across the human genome, altering the epigenetic landscape of several cellular genes. Overall, our data indicate that EBV utilizes PARP1 to induce both viral and human epigenetic programs that can sustain EBV persistent infection.
PARP Pol
CTCF-PARlyation
PARP1 binds to EBV genome during latency (Lupercalia-Green et al JV 2018)
(Cool should check this up, there are peaks!)
PARP1 inhibitionalters CTCF binding. (+ Olaparib)
- PARP1 inhibition alters CTCF binding
- Represses Cp promoter activity (Cp-Type III, QP -Type I)
EBV adopts alternative chromosome conformation
LMP2 regulates EBV genome chromatin structure through PARP1
PRPPi (olaparib) offsets LMP10mediatd gene activation
- Targets are involved in cell metabolism
LMP1 expression reorganizes the epigenomic compartments of hoist genome
- By 3C-Hi contact
- AB compartment switch ~ 200kb resolution
- LMP1 regulates both host and viral genomes chromatin structure through PARP1
6:00 PM – 11:00 PM Closing Banquet Great Hall