由 sufang 在 日, 10/26/2014 – 09:25 發表 Pre-published IDO1 TDO2
Time: 10:00~11:30 October 24, 2014 (Fri)
Topic: IDO pathways in pathogenic inflammation and immune escape in cancer
Speaker: George C. Prendergast, PhD (CV download)
Professor, President & CEO,
Lankenau Institute for Medical Research
Editor-in-Chief, Cancer Research (他說兩大要件: (1) Pathological relevalence (2) Translational relevance. 若是一堆western blot的,他建議送”Cancer Cell”, 一堆signaling pathway的,他會轉送Molecular Cancer Research)
2013年秋天,該講者在緬因州MDI Biological Laboratory也有一場「講得飛快」的Webnair (YouTube).
記下一些我聽得懂的keyword and reference, 不對的地方請按右下角「回應」指正與補充, 謝謝.
1. 許多Tumor cell模仿懷孕中之胎盤 (Science 1998),透過大量表現IDO1,迅速分解靠過來的T cell細胞中之tryptophan分解,造成T cell growth arrest. (參考資料 Nat Med 2003 Vol9, pmid: 14502282)
補充一: 該篇文章檢查哪些癌細胞株中 IDO1 mRNA is constitiutively-on, 其中有一株是pancreatic carcinoma, 一株laryngeal carcinoma, 一株pharyngeal SCC 原來十一年前就註定要與IDO相遇?)
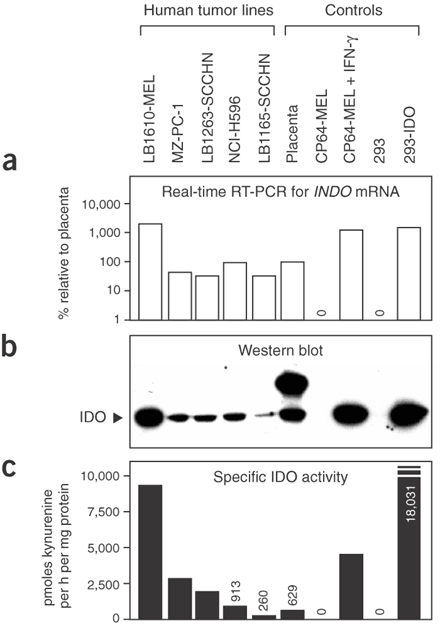

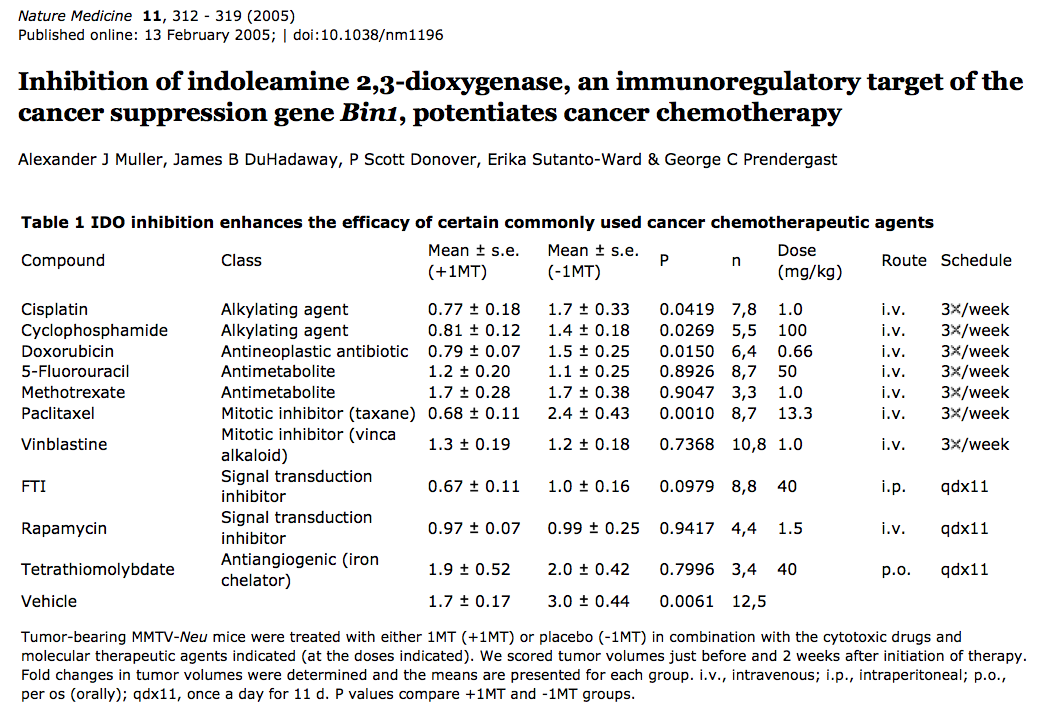
補充二、Discussion第一段: IDO is a cytosolic enzyme, so tryptophan degradation by IDO occurs inside the cell. Because tryptophan readily crosses the plasma membrane through specific transporters, a microenvironment depleted of tryptophan is created in the vicinity of IDO-expressing cells, which function as ‘tryptophan sinks’. Protein synthesis proceeds despite lowered levels of tryptophan, presumably because the Km of tryptophanyl-tRNA synthetase for tryptophan is lower than that of IDO. This enables most cells to maintain their growth in the presence of IDO. For instance, although tumor cells expressing high levels of IDO have a reduced growth rate in vitro, their proliferation is not arrested. T lymphocytes, in contrast, stop proliferating under such conditions because they have a tryptophan-sensitive checkpoint, which blocks their cell cycle in the G1 phase when tryptophan concentration is below 0.5−1 uM (真是有許許多多的「米角」)
2. 最早是在Bin1 loss的老鼠中發現IDO1大量表現 (Nat Med 2005, pmid: 15711557),講者推測是微量的nulcear Bin1貢獻,真正mechanism為何,目前仍不是很清楚。
補充: Official Symbol BIN1 (Official Full Name bridging integrator 1)
Also known as AMPH2; AMPHL; SH3P9
Summary: This gene encodes several isoforms of a nucleocytoplasmic adaptor protein, one of which was initially identified as a MYC-interacting protein with features of a tumor suppressor. Isoforms that are expressed in the central nervous system may be involved in synaptic vesicle endocytosis and may interact with dynamin, synaptojanin, endophilin, and clathrin. Isoforms that are expressed in muscle and ubiquitously expressed isoforms localize to the cytoplasm and nucleus and activate a caspase-independent apoptotic process. Studies in mouse suggest that this gene plays an important role in cardiac muscle development. Alternate splicing of the gene results in ten transcript variants encoding different isoforms. Aberrant splice variants expressed in tumor cell lines have also been described. [provided by RefSeq, Sep 2011]
Also known as AMPH2; AMPHL; SH3P9
Summary: This gene encodes several isoforms of a nucleocytoplasmic adaptor protein, one of which was initially identified as a MYC-interacting protein with features of a tumor suppressor. Isoforms that are expressed in the central nervous system may be involved in synaptic vesicle endocytosis and may interact with dynamin, synaptojanin, endophilin, and clathrin. Isoforms that are expressed in muscle and ubiquitously expressed isoforms localize to the cytoplasm and nucleus and activate a caspase-independent apoptotic process. Studies in mouse suggest that this gene plays an important role in cardiac muscle development. Alternate splicing of the gene results in ten transcript variants encoding different isoforms. Aberrant splice variants expressed in tumor cell lines have also been described. [provided by RefSeq, Sep 2011]
3. 接著,他show了幾張2008年他寫在Oncogene的一篇review上的圖,包括IDO1也會由MDSC, APC等細胞表現。
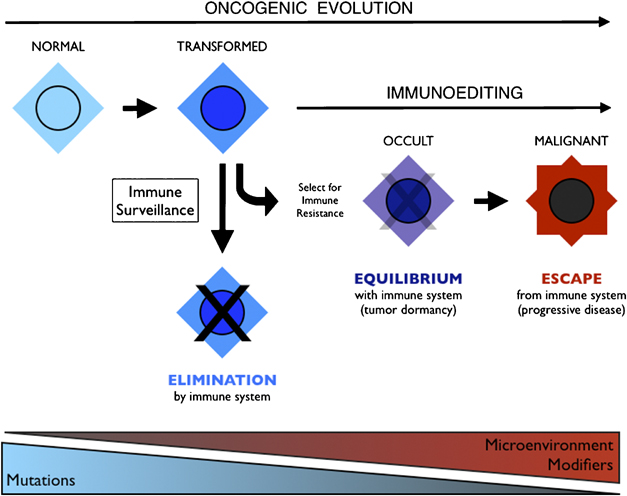
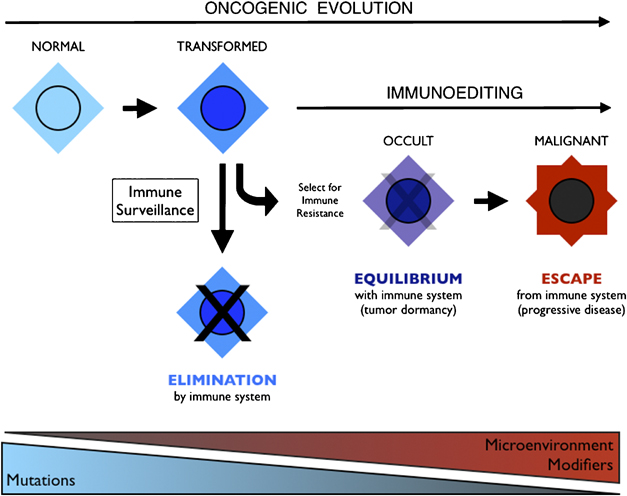
Integration of immunoediting and oncogenesis during cancer progression. Oncogenesis initiates the formation of transformed cells that are attacked by immune cells as the result of neoantigen presentation. This process of immune surveillance imposes a selection for transformed cells that acquire tactics to escape control. Genetic instability driven by oncogenesis facilitates evolution of strategies for immune evasion or suppression, the latter of which may help tilt the tumor microenvironment from hostile to supportive for tumor cells. As the battle evolves between tumor cells and the immune system, representing the process of immunoediting, a state of immune equilibrium may be achieved that corresponds to a dynamic but dormant disease that is clinically occult. Further iteration of evasion mechanisms to defeat different elements of the immune system may ultimately drive immune suppression beyond the local microenvironment, achieving immune escape and thereby licensing invasive and metastatic behaviors that define progressive disease. Invasion and metastasis may be derivative of immune escape since the former are presumably impotent while immune surveillance prevails beyond the local microenviroment. While mutations may initiate cancer, modifiers and microenvironmental factors including those sculpted by the process of immunoediting may dictate the course of disease.
 IDO (indoleamine 2,3-dioxygenase) and IDO2 control a tryptophan catabolism signaling pathway. (a) From tryptophan starvation to LIP activation. By catabolizing the essential amino acid tryptophan, IDO and IDO2 generate kynurenines and other reaction products that can modulate T-cell immunity as well as a local microenvironment that is starved for tryptophan. Little is known as yet of the precise mechanistic effects of the tryptophan catabolites generated. Elaboration of the starvation condition triggers a stress response in local T cells through Gcn2, which responds to amino-acid deprivation by phosphorylating the translation initiation factor eIF2alpha, leading to a blockade of most translation initiation with the exception of certain factors such as LIP involved in mediating responses to the stress. (b) LIP is a dominant negative isoform of the immune regulatory b/ZIP transcription factor NF-IL6, also known as CEBPbeta. LIP is an alternately translated isoform of the transcription factor NF-IL6/CEBPbeta implicated in regulating proliferation and immune response. Starvation responses switch NF-IL6/CEBPbeta expression from LAP isoforms to the LIP isoform through the use of a downstream translation start site in the mRNA. Encoding only a b/ZIP dimerization domain, LIP functions as a ‘natural’ dominant negative molecule that disrupts NF-IL6/CEBPbeta function by competing with LAP isoforms for binding to target gene promoters. Both IDO and IDO2 can switch on LIP, but subsequent restoration of tryptophan levels will only switch it off in the case of IDO, offering a possible mechanism for distal propagation of immune suppression away from the local tumor microenvironment (Figure 5). NF-IL6/CEBPbeta target genes with relevance to the function of IDO include the immune suppressive cytokines IL-6, IL-10 and TGF-beta, which may be upregulated as a result of LIP induction.
IDO (indoleamine 2,3-dioxygenase) and IDO2 control a tryptophan catabolism signaling pathway. (a) From tryptophan starvation to LIP activation. By catabolizing the essential amino acid tryptophan, IDO and IDO2 generate kynurenines and other reaction products that can modulate T-cell immunity as well as a local microenvironment that is starved for tryptophan. Little is known as yet of the precise mechanistic effects of the tryptophan catabolites generated. Elaboration of the starvation condition triggers a stress response in local T cells through Gcn2, which responds to amino-acid deprivation by phosphorylating the translation initiation factor eIF2alpha, leading to a blockade of most translation initiation with the exception of certain factors such as LIP involved in mediating responses to the stress. (b) LIP is a dominant negative isoform of the immune regulatory b/ZIP transcription factor NF-IL6, also known as CEBPbeta. LIP is an alternately translated isoform of the transcription factor NF-IL6/CEBPbeta implicated in regulating proliferation and immune response. Starvation responses switch NF-IL6/CEBPbeta expression from LAP isoforms to the LIP isoform through the use of a downstream translation start site in the mRNA. Encoding only a b/ZIP dimerization domain, LIP functions as a ‘natural’ dominant negative molecule that disrupts NF-IL6/CEBPbeta function by competing with LAP isoforms for binding to target gene promoters. Both IDO and IDO2 can switch on LIP, but subsequent restoration of tryptophan levels will only switch it off in the case of IDO, offering a possible mechanism for distal propagation of immune suppression away from the local tumor microenvironment (Figure 5). NF-IL6/CEBPbeta target genes with relevance to the function of IDO include the immune suppressive cytokines IL-6, IL-10 and TGF-beta, which may be upregulated as a result of LIP induction.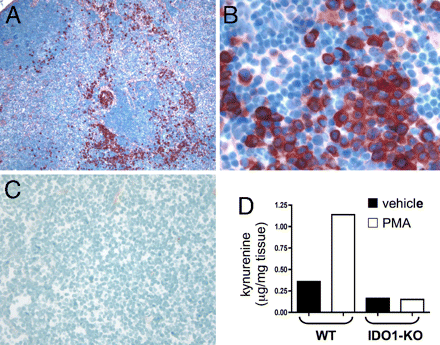
Topical PMA treatment stimulates IDO1 expression in dLNs. (A–C) Inguinal dLN sections from PMA-treated (A and B) and untreated B6 (C) mice were stained with anti-IDO Ab (red chromogen; magnifications, 100x in A and C, 400x in B). (D) Kynurenine present in homogenized dLNs from PMA and acetone (vehicle) treated BALB/c (WT) and IDO1-KO mice was measured by LC/MS/MS as described in Methods.
5. 2010年Muller et al 這一篇和Am J Pathol這一篇繼續闡述PNAS的工作,強調”IDO programs a cancerous environment = inflammation = immune escape”
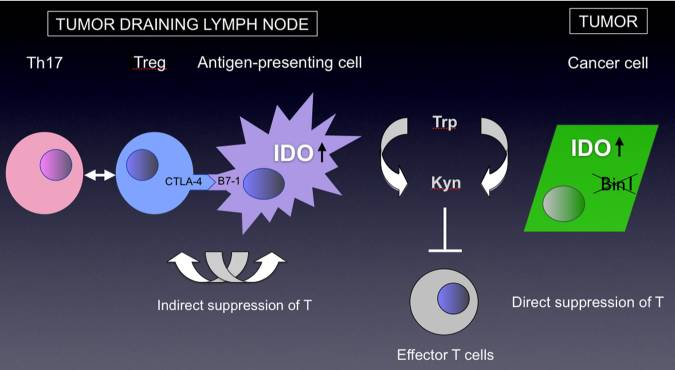
Dual roles of IDO in cancer. IDO is overexpressed in many types of tumor cells where its presence has been associated with poor prognosis. In tumor cells, IDO dysregulation has been linked to inactivation of tumor suppressor gene Bin1, an amphiphysin-like gene that is widely attenuated or misspliced in cancer. In tumor-draining lymph nodes, IDO is upregulated in a regulatory subset of plasmacytoid dendritic cells that has been suggested to inform a positive feedback loop in the activation of T regulatory cells (Tregs), while also blunting their interconversion to Th17 inflammatory cells. IDO activity consumes tryptophan, depriving it from local tissue microenvironments, also generating kynurenine as a product. Both of these processes have been implicated in T cell suppression, including by blunting T effector cell function as well as recruitment of Tregs.

DO functions primarily in non-hematopoietic cells to support Inflammatory skin carcinogenesis
a Experimental design. Four-week-old recipient mice were lethally irradiated and reconstituted with bone marrow from 6-week-old donor mice. All control and reciprocal transplant possibilities in wild-type and Ido1−/− mice were performed. After allowing 6 weeks for the transplanted bone marrow to repopulate the hematopoietic system, host mice were subjected to two-stage Inflammatory skin carcinogenesis. b Papilloma formation in bone marrow chimeric mice. Legend indicates host-to-donor configuration in each cohort (n = 10). Standard error is indicated for each of the incidence curves. KO, Ido1−/− mice.
6. 2012年 Cancer Discov提及IDO和Lung cancer metastasis之關聯
RAS/MEK -> IDO ->IL6 -> MDSC -> immune escape
IDO deficiency mice unexpectedly exhibited an angiogenic defect!
Abstract
Indoleamine 2,3-dioxygenase (IDO) enzyme inhibitors have entered clinical trials for cancer treatment based on preclinical studies, indicating that they can defeat immune escape and broadly enhance other therapeutic modalities. However, clear genetic evidence of the impact of IDO on tumorigenesis in physiologic models of primary or metastatic disease is lacking. Investigating the impact of Ido1 gene disruption in mouse models of oncogenic KRAS-induced lung carcinoma and breast carcinoma–derived pulmonary metastasis, we have found that IDO deficiency resulted in reduced lung tumor burden and improved survival in both models. Micro-computed tomographic (CT) imaging further revealed that the density of the underlying pulmonary blood vessels was significantly reduced in Ido1-nullizygous mice. During lung tumor and metastasis outgrowth, interleukin (IL)-6 induction was greatly attenuated in conjunction with the loss of IDO. Biologically, this resulted in a consequential impairment of protumorigenic myeloid-derived suppressor cells (MDSC), as restoration of IL-6 recovered both MDSC suppressor function and metastasis susceptibility in Ido1-nullizygous mice. Together, our findings define IDO as a prototypical integrative modifier that bridges inflammation, vascularization, and immune escape to license primary and metastatic tumor outgrowth.
Significance: This study provides preclinical, genetic proof-of-concept that the immunoregulatory enzyme IDO contributes to autochthonous carcinoma progression and to the creation of a metastatic niche. IDO deficiency in vivo negatively impacted both vascularization and IL-6–dependent, MDSC-driven immune escape, establishing IDO as an overarching factor directing the establishment of a protumorigenic environment.
Introduction
Inflammatory tissue microenvironments contribute strongly to tumor progression, but due to the complex multifactorial nature of inflammation, there remains limited understanding of specific pathogenic components that might be targeted to effectively treat cancer (1). In this context, the tryptophan-catabolizing enzyme indoleamine 2,3-dioxygenase (IDO) has emerged as an intriguing target implicated in tumoral immune escape (2, 3). IDO-inhibitory compounds have entered clinical trials based on evidence of immune-based antitumor responses in a variety of preclinical models of cancer (4–4). Meanwhile, inadvertent IDO targeting may already be providing benefits to patients as illustrated by recent evidence that the clinically approved tyrosine kinase inhibitor imatinib dampens IDO induction as a key mechanism for achieving therapeutic efficacy in the treatment of gastrointestinal stromal tumors (11).
Although results with IDO pathway inhibitors are provocative, the conclusions that can be drawn are inherently limited by drug specificity concerns, especially in the absence of independent genetic validation. Addressing this issue, our studies on the impact of Ido1 gene deletion on 7,12-dimethylbenz(a)anthracene/12-O-tetradecanoylphorbol-l3-acetate (DMBA/TPA)- elicited skin papillomagenesis established that IDO has an integral tumor-promoting role in the context of phorbol ester–elicited inflammation (12, 13), but interpretation of these results is tempered by the possibility that the chemical exposures in this model may produce anomalies irrelevant to the majority of spontaneous tumors. The lungs present a particularly compelling physiologic context in which to further investigate the role of IDO in tumorigenesis as IDO is known to be highly inducible in this tissue (14, 15), and there is an urgent unmet medical need for effective therapeutic options to treat primary lung tumors and metastases. In this report, we investigated the consequences of IDO loss through genetic ablation in the context of well-established, pulmonary models of oncogenic KRAS-induced adenocarcinoma and orthotopic breast carcinoma metastasis. Our findings reveal previously unappreciated roles for IDO in vascularization and in the production of the proinflammatory cytokine interleukin (IL)-6 that in turn dictates the development of protumorigenic, myeloid-derived suppressor cells (MDSC).
7. 2012年Oncoimmunlogy提及IDO-mTOR之關聯、mTOR and D-1MT (=Indoximod=NLG8189)

Figure 6. Mechanistic model. Trp deprivation caused by IDO generates signals sensed by distinct amino acid sufficiency and deficiency pathways. Trp deficiency is sensed by the integrated stress kinase GCN2 that inhibits eIF-2α and blocks translation. Through a distinct pathway, the lack of Trp sufficiency causes mTOR to be inactivated, leading to autophagy via LC3 de-repression and translational blockade via S6K inactivation. D-1MT acts as a mimetic of Trp in the sufficiency pathway, thereby functionally reversing the effects of IDO on mTOR and autophagy and potentially Treg formation controlled by PKC-Θ.
8. 2011年Nat Med提及imatinib-treated GIST其實一部份是因為降低KIT-induced IDO expression (主文).
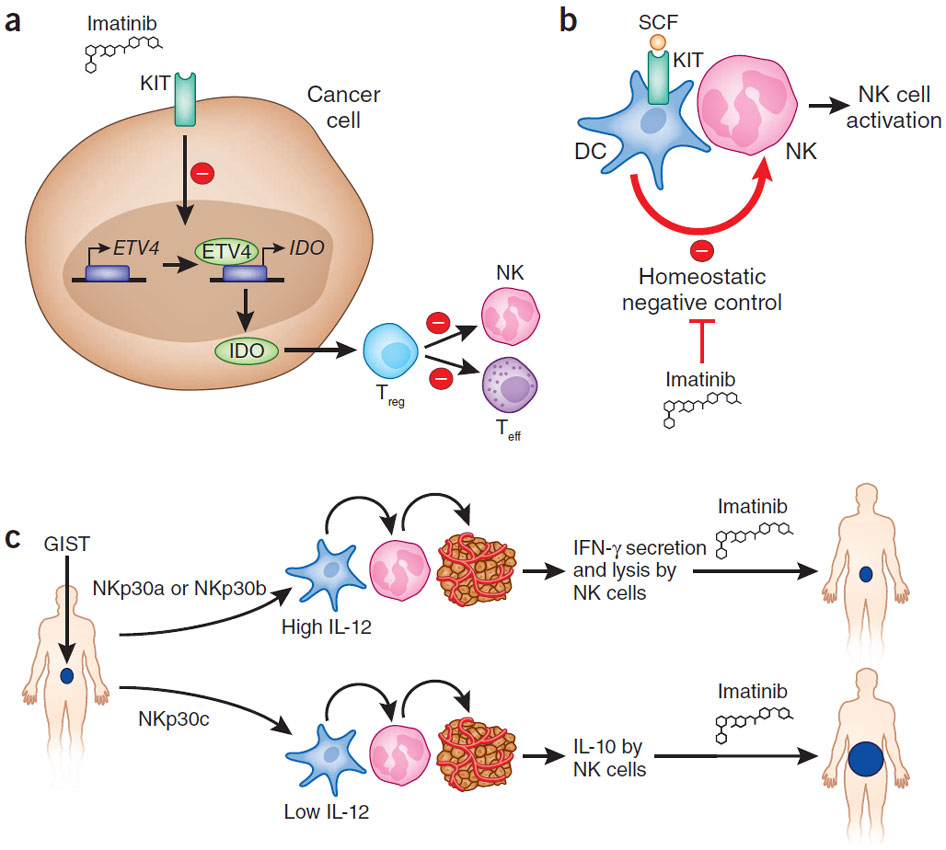
a) Suppression of oncogene addiction. Balachandran et al.3 show that activated KIT induces expression of the transcription factor ETV4, which transactivates IDO. IDO converts tryptophan into its immunosuppressive metabolites within the tumor microenvironment, stimulating Treg cells, resulting in the local inhibition of CD8+ cytotoxic T lymphocytes and NK cells7. As a result, oncogene addiction is directly linked to local immunosuppression. Inhibition of KIT by imatinib reverts this state. Teff, effector T cells. (b) Priming of NK cells by imatinib-treated DCs. Imatinib interferes with KIT signaling in immature DCs, promoting DC-mediated NK cell activation5. (c) NK cell recognition of dendritic or tumor cells according to NKp30 profiling. Individuals presenting with an NKp30a or NKp30b profile harbor NK cells that secrete IFN-γ when they encounter tumor or DCs, which in turn, then produce interleukin (IL)-12. In contrast, in these conditions, NKs and DCs from individuals with an NKp30c profile produce IL-10 and little IL-12, respectively. SCF, stem cell factor.
9.有無pathological IDO1 SNPs? Dr. Prendergast回說他們做過大概1%的機率在IDO1 promoter region有個functional SNP. IDO2 protein則有兩個。
#1由 sufang 在 日, 10/26/2014 – 10:13 發表。
瘋狂的想法…
Humanized mouse model for oral cancer/IDO/TDO is the way to go!!
#2由 sufang 在 日, 10/26/2014 – 09:48 發表。
擷自NICRLINE_安靜版 2014/10/25 (六)
10:00 素芳@pfi Jonshon, 有件事要麻煩你查一下、40對T/N expression array中 INDO (=IDO1), TDO2都有signal, 但INDOL1 (=IDO2)卻找不到,可以追一下是從頭到尾Illumina chip上就沒有這個probe? 還是在normalization過程中被踢走了?
10:12 江士昇 Ok
10:13 素芳@pfi IDO1, IDO2似乎side-by-side座落在chromosome 8 上面, 昨天演講後、去樓上與生藥所round table discussion時, Dr. Pendergast 說這兩個enzyme結構、功能似乎很重複,在表現調節上,是透過alternative splicing在調控.TDO2則是和它們不一樣的enzyme, 生藥所伍素瑩老師也有提到一點TDO2的事情,這方面應該再去請教她.(因為石所長有問Dr. Prendergast說what’s the next? 講這回答說: maybe the combination of IDO/TDO inhibitors)
10:18 素芳@pfi 我有跟他分享說我們40對檢體TDO2 transcript百分百在tumor part上揚,IDO1 80%,但IDO2卻完全silence. 回來想一想,咦,IDO2是沒變化,還是根本測不到? 所以我翻回11SVA那張excel, 但遍尋不到INDOL1的record. 謝謝Johnson!!
10:13 素芳@pfi IDO1, IDO2似乎side-by-side座落在chromosome 8 上面, 昨天演講後、去樓上與生藥所round table discussion時, Dr. Pendergast 說這兩個enzyme結構、功能似乎很重複,在表現調節上,是透過alternative splicing在調控.TDO2則是和它們不一樣的enzyme, 生藥所伍素瑩老師也有提到一點TDO2的事情,這方面應該再去請教她.(因為石所長有問Dr. Prendergast說what’s the next? 講這回答說: maybe the combination of IDO/TDO inhibitors)
10:18 素芳@pfi 我有跟他分享說我們40對檢體TDO2 transcript百分百在tumor part上揚,IDO1 80%,但IDO2卻完全silence. 回來想一想,咦,IDO2是沒變化,還是根本測不到? 所以我翻回11SVA那張excel, 但遍尋不到INDOL1的record. 謝謝Johnson!!